NuPoP update
NuPoP is an R package for Nucleosome Positioning Prediction based on DNA sequence. The model is trained based on in vivo chemical map or Mnase map of nucelosomes.
Nucleosome is the foundamental packing unit of DNA in eukaryotic cells.
Its positioing is largely determined by intrinsic code in the DNA sequence. Nucleosome positioning regulates varous of cell activities such as transcription by controlling accessibity of protein factor binding. See update at the github site below.
BoostMEC new
BoostMEC is a boosting tree based approach, utilizing LightGBM for the prediction of wild-type CRISPR-Cas9 editing efficiency.
CRISPR-Cas9 is the cutting-edge genome editting techonology. A variety of sgRNA properties have been found to be predictive of CRISPR cleavage efficiency, including the position-specific sequence composition of sgRNAs, global sgRNA sequence properties, and thermodynamic features. BoostMEC maintains an advantage over other state-of-the-art CRISPR efficiency prediction models that are based on deep learning through its ability to produce more interpretable feature insights and predictions.
DNAcycP new
DNAcycP has a Python package and a web server for DNA cyclizability or bendability prediction trained based on loop-seq data.
DNA mechanical properties play a critical role in every aspect of DNA-dependent biological processes. Recently a high throughput assay named loop-seq has been developed to quantify the intrinsic bendability of a massive number of DNA fragments simultaneously. DNAcycP, built on a deep-learning approach, predicts intrinsic DNA cyclizability with high fidelity compared to the experimental data.
SPECIES

SPECIES is an R package that implements various popular methods in species richness estimation.
How many butterfly species are there in a ecosystem? What was the vocabulary size of Shakespere? How many microbial species are there in human's gut system? All of these questions are related to a problem known as species number estimation problem.
RiboDiPA
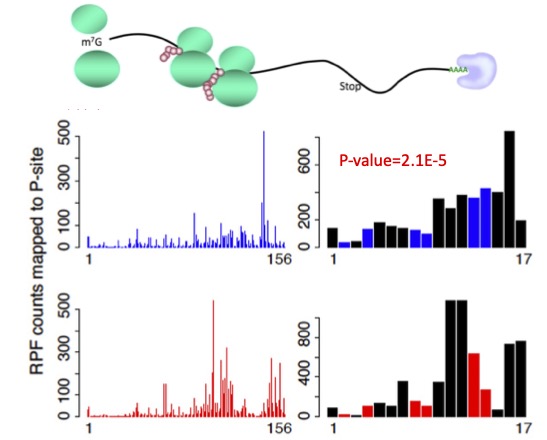
RiboDiPA is a bioinformatics pipeline developed for differential pattern analysis of Ribo-seq footprint data.
Ribosome is a protein complex that binds to mRNA sequence to facilitate translation. Its footprint, collected by ribo-seq assay, provides a snapshot of translation activities in cells in vivo.
DegNorm
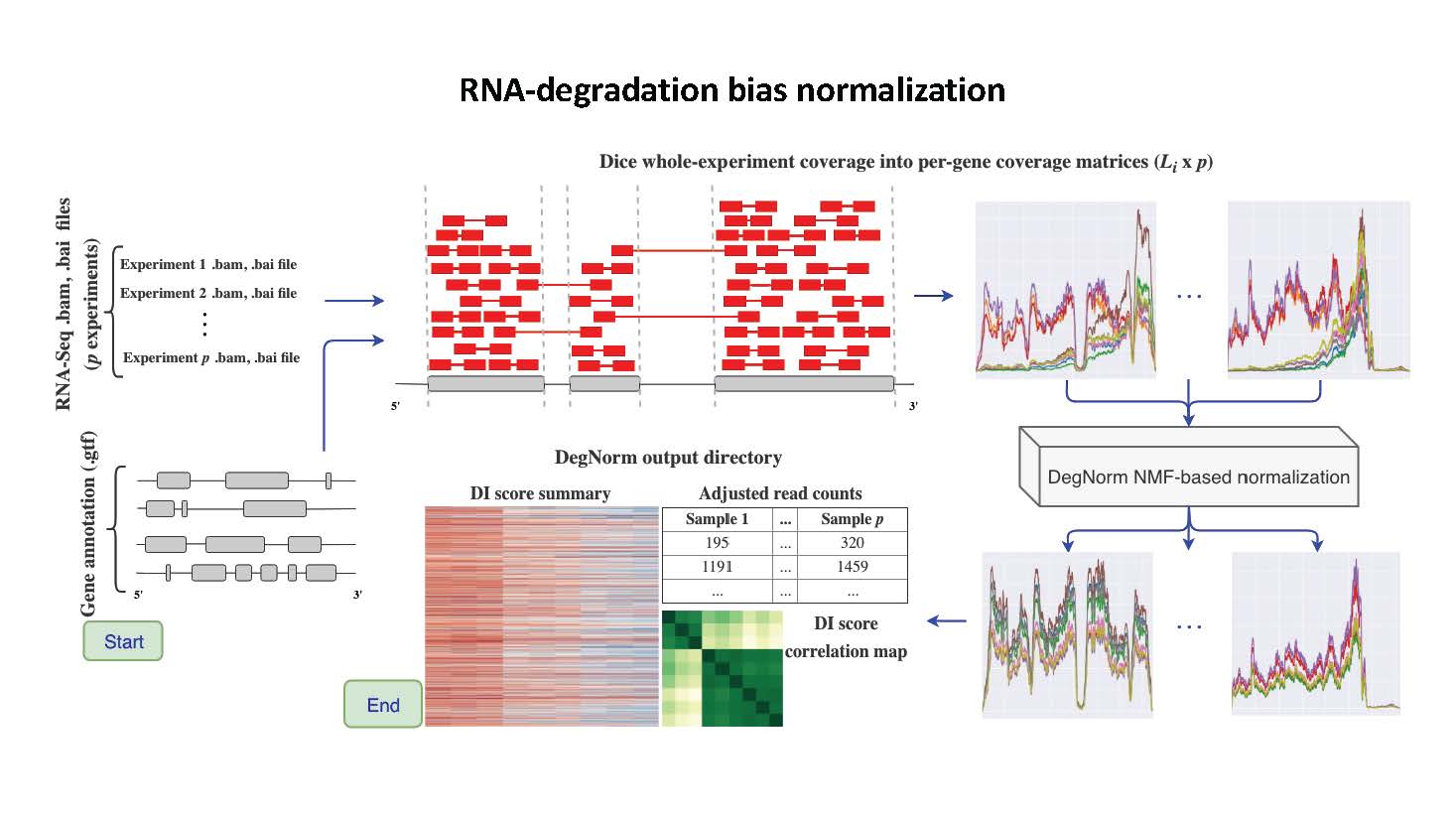
DegNorm is a bioinformatics pipeline to correct for bias due to the heterogeneous patterns of transcript degradation in RNA-seq data.
RNA degradation is a commonly used measure for sample quality in RNA-seq, which is sample and gene-specific. DegNorm provides a sample- and gene-specific correction of bias due to degradation.

